People
Patricia Reggio

Field: Computational Chemistry and Computer-Aided Drug Design
Room: 406 Sullivan Science Building
Phone: 336.334.5333
Email: phreggio@uncg.edu
Research Website: https://chem.uncg.edu/person/patricia-reggio/
Education
B.S., Chemistry, Louisiana State University at New Orleans,1971
Ph.D., Physical Chemistry, University of New Orleans,1978
Postdoctoral Fellow, University of New Orleans,1979
Research
I have always been interested in understanding how things work at the molecular level. The major focus of my research group has been on the G protein-coupled cannabinoid receptors for many years. This includes the well-known cannabinoid CB1 and CB2 receptors, as well as new orphan receptors that appear to be cannabinoid receptors, GPR55 and GPR18. My group has been very fortunate to have funding from the National Institutes of Health/National Institute on Drug Abuse for many years. We are also fortunate to have many research collaborators who bring interesting new questions and problems for us to help solve (one of which I discuss below). Technically, we are a computational chemistry group, so we have experience with biomolecular simulations of large systems (such as ligands traveling to and activating their receptors and activated receptors sending their messages via signaling proteins, such as G-proteins or beta-arrestins). We are also interested in designing drugs for a specific target using computational chemistry and optimizing them for use as a medication. Our calculations are carried out on a large, new GPU-enabled Linux cluster that is part of the Biomolecular Simulation and Bioinformatics Core Lab at UNCG. To do all that we do, we have to read widely to have a good grasp of protein structure, biophysics, medicinal chemistry, receptor pharmacology and biochemistry. So students in my group read a lot of scientific literature, work hard, present their work at national and international scientific meetings and hopefully learn a lot. Below is a brief summary of some current projects in my lab, including a few projects that originated from interesting questions we were asked to help solve.
Structural Basis of CB2 Receptor-Gi Protein Interaction
Work Done by Jagjeet S. Mnoptra, PhD Student
Supported by National Institutes of Health Grants (DA003934 and DA021358)
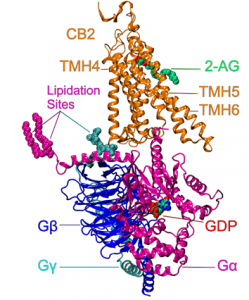 Physiologically, GPCRs are activated by ligands (extracellular or membrane based) that enable the receptors to interact with and activate distinct sets of heterotrimeric G proteins (Gαβγ), as well as b-arrestins. We recently used our CB2 receptor model that had been activated by the endogenous ligand, 2-AG to study the complex that is formed with G-protein upon receptor activation using all-atom molecular dynamics (MD). This project was performed by my PhD student, Jagjeet S. Mnpotra. We used experimental crosslinking results for CB2 with Gi from Dr. Zhao-Hui Song’s lab at University of Louisville to validate the simulations. Our results showed that although our MD simulations started with the G protein orientation in the β2-AR*/ Gαsβ1γ2 complex crystal structure, the Gαi1β1γ2 protein re-oriented itself within 300 ns. Two major changes occurred: (1) The Gαi1 α5 helix tilt changed due to the outward movement of TMH5 in CB2 R*. (2) A 25 degree clockwise rotation of Gαi1β1γ2 underneath CB2 R* occurred, with rotation ceasing when P139 (IC2 loop) anchors in a hydrophobic pocket on Gαi1 (V34, L194, F196, F336, T340, I343 and I344). In this complex, all three experimentally identified crosslinks can occur. These findings should be relevant for other Class A GPCRs that couple to Gi proteins.Physiologically, GPCRs are activated by ligands (extracellular or membrane based) that enable the receptors to interact with and activate distinct sets of heterotrimeric G proteins (Gαβγ), as well as b-arrestins. We recently used our CB2 receptor model that had been activated by the endogenous ligand, 2-AG to study the complex that is formed with G-protein upon receptor activation using all-atom molecular dynamics (MD). This project was performed by my PhD student, Jagjeet S. Mnpotra. We used experimental crosslinking results for CB2 with Gi from Dr. Zhao-Hui Song’s lab at University of Louisville to validate the simulations. Our results showed that although our MD simulations started with the G protein orientation in the β2-AR*/ Gαsβ1γ2 complex crystal structure, the Gαi1β1γ2 protein re-oriented itself within 300 ns. Two major changes occurred: (1) The Gαi1 α5 helix tilt changed due to the outward movement of TMH5 in CB2 R*. (2) A 25 degree clockwise rotation of Gαi1β1γ2 underneath CB2 R* occurred, with rotation ceasing when P139 (IC2 loop) anchors in a hydrophobic pocket on Gαi1 (V34, L194, F196, F336, T340, I343 and I344). In this complex, all three experimentally identified crosslinks can occur. These findings should be relevant for other Class A GPCRs that couple to Gi proteins. In addition to Dr. Song, we collaborated with Dr. Mike Pitman at IBM, Dr. Alan Grossfield at University of Rochester and Dr. Klaus Gawrisch at NIAAA on this project as well.
Physiologically, GPCRs are activated by ligands (extracellular or membrane based) that enable the receptors to interact with and activate distinct sets of heterotrimeric G proteins (Gαβγ), as well as b-arrestins. We recently used our CB2 receptor model that had been activated by the endogenous ligand, 2-AG to study the complex that is formed with G-protein upon receptor activation using all-atom molecular dynamics (MD). This project was performed by my PhD student, Jagjeet S. Mnpotra. We used experimental crosslinking results for CB2 with Gi from Dr. Zhao-Hui Song’s lab at University of Louisville to validate the simulations. Our results showed that although our MD simulations started with the G protein orientation in the β2-AR*/ Gαsβ1γ2 complex crystal structure, the Gαi1β1γ2 protein re-oriented itself within 300 ns. Two major changes occurred: (1) The Gαi1 α5 helix tilt changed due to the outward movement of TMH5 in CB2 R*. (2) A 25 degree clockwise rotation of Gαi1β1γ2 underneath CB2 R* occurred, with rotation ceasing when P139 (IC2 loop) anchors in a hydrophobic pocket on Gαi1 (V34, L194, F196, F336, T340, I343 and I344). In this complex, all three experimentally identified crosslinks can occur. These findings should be relevant for other Class A GPCRs that couple to Gi proteins.Physiologically, GPCRs are activated by ligands (extracellular or membrane based) that enable the receptors to interact with and activate distinct sets of heterotrimeric G proteins (Gαβγ), as well as b-arrestins. We recently used our CB2 receptor model that had been activated by the endogenous ligand, 2-AG to study the complex that is formed with G-protein upon receptor activation using all-atom molecular dynamics (MD). This project was performed by my PhD student, Jagjeet S. Mnpotra. We used experimental crosslinking results for CB2 with Gi from Dr. Zhao-Hui Song’s lab at University of Louisville to validate the simulations. Our results showed that although our MD simulations started with the G protein orientation in the β2-AR*/ Gαsβ1γ2 complex crystal structure, the Gαi1β1γ2 protein re-oriented itself within 300 ns. Two major changes occurred: (1) The Gαi1 α5 helix tilt changed due to the outward movement of TMH5 in CB2 R*. (2) A 25 degree clockwise rotation of Gαi1β1γ2 underneath CB2 R* occurred, with rotation ceasing when P139 (IC2 loop) anchors in a hydrophobic pocket on Gαi1 (V34, L194, F196, F336, T340, I343 and I344). In this complex, all three experimentally identified crosslinks can occur. These findings should be relevant for other Class A GPCRs that couple to Gi proteins. In addition to Dr. Song, we collaborated with Dr. Mike Pitman at IBM, Dr. Alan Grossfield at University of Rochester and Dr. Klaus Gawrisch at NIAAA on this project as well.
Where Does Pregnenolone, an Endogenous Allosteric Modulator of the Cannabinoid CB1 Receptor, Bind and How Does It Work?
Work done by Dow P. Hurst, Research Scientist
Supported by National Institutes of Health Grants (DA003934 and DA021358)
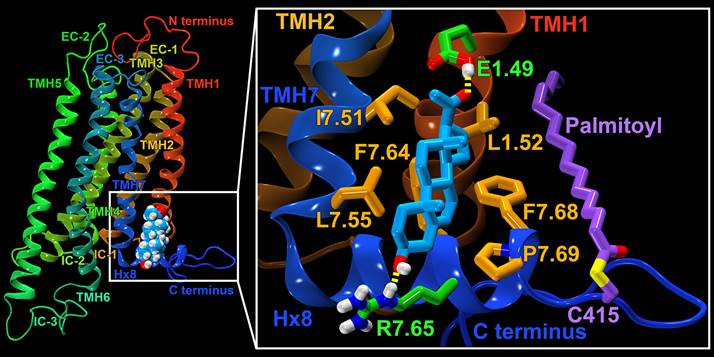
It is now recognized that many GPCRs contain allosteric binding sites for endogenous and/or synthetic ligands, which are topographically distinct from the agonist-binding site (the orthosteric site). In contrast to the direct effects on receptor function that are mediated by orthosteric ligands, allosteric drugs act by modulating receptor activity through conformational changes in the receptor that are transmitted from the allosteric to the orthosteric site and/or to effector coupling sites.
Pregnenolone is a precursor of cholesterol that until recently was thought not to act as a specific agent itself. Our Research Collaborator, Dr. Pier-Vincenzo Piazza at INSERM in France discovered that the administration of the principal psychoactive ingredient in Cannabis sativa (marijuana), ∆9-tetrahydrocannabinol (THC), substantially increases the synthesis of pregnenolone in the brain via the activation of the CB1 receptor. Pregnenolone then, acting as a signalling specific inhibitor of the CB1 receptor, reduces the intoxicating effects of THC. This negative feedback mediated by pregnenolone reveals an unknown paracrine/autocrine loop protecting the brain from CB1 receptor over-activation that could open an unforeseen novel approach for the treatment of cannabis intoxication and addiction.
Question To Us: We were asked to figure out where pregnenolone binds at the CB1 receptor. To identify specific binding site(s) for pregnenolone binding at CB1, we used the Forced-Biased Metropolis Monte Carlo simulated annealing program (MMC). The receptor protein was immersed in a simulated field of four fragments (two polar fragments and two non-polar fragments) derived from the pregnenolone structure. The chemical potential was then annealed by lowering the system chemical potential, causing only those fragments with the best free energy of binding to the protein surface to remain. While each fragment bound to multiple positions on the CB1 receptor, there was only one region in which all four fragments clustered in the correct spatial proximity. This was the transmembrane helix 1 (TMH1)/TMH7/Hx 8 lipid facing region in CB1, where a polar residue (E1.49) is located on the lipid face of CB1 near the midpoint of the TMH region (see figure above). Receptor E1.49G mutation studies supported our site identification. We were also able to suggest that because of the location of the pregnenolone binding site (which constrains the movement of TMH1 and TMH7), pregnenolone should inhibit some, but not all effects of THC. This is in fact what was seen experimentally.
Allosteric Modulation of the Cannabinoid CB1 Receptor: Binding Site Elucidation and Relationship to G-Protein Signaling
Work done by Derek M. Shore, PhD Student
Supported by National Institutes of Health Grants (DA003934 and DA021358)
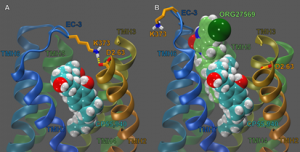 Another current project in my lab is looking at the allosteric modulator, ORG27569 and its effects on the affinity and signalling of the cannabinoid agonist, CP55,940. The goal of this work was to identify ORG27569’s binding site at CB1. To this end, we used computational, synthesis, mutation, and functional studies to identify ORG27569’s binding site in the CB1 TMH3-6-7 region. This site is consistent with the results of K3.28192A, F3.36200A, W5.43279A, W6.48356A, and F3.25189A mutation studies, which revealed ORG27569’s binding site overlaps with our previously determined binding site of SR141716A, but extends extracellularly. Additionally, we identified a key electrostatic interaction between ORG27569’s piperidine ring nitrogen and K3.28192 that is important for ORG27569 to act as an inverse agonist. At this allosteric site, ORG27569 promotes an intermediate conformation of the CB1 receptor, explaining ORG27569’s ability to increase CP55,940’s equilibrium binding. This site also explains ORG27569’s ability to antagonize CP55,940’s efficacy in three complimentary ways: (1) ORG27569 sterically blocks movements of the second extracellular loop that have been linked to receptor activation; (2) ORG27569 sterically blocks a key electrostatic interaction between the third extracellular loop residue K373 and D2.63176; and (3) ORG27569 packs against TMH6, sterically hindering movements of this helix that have been shown to be important for receptor activation. My PhD student, Derek Shore, developed the model of the ORG binding site. Compound synthesis was performed by Dr. Herb Seltzman at Research Triangle Institute, mutation studies were performed by Dr. Mary Abood at Temple University and pharmacological studies were performed by Dr. Ruth Ross at University of Toronto.
Another current project in my lab is looking at the allosteric modulator, ORG27569 and its effects on the affinity and signalling of the cannabinoid agonist, CP55,940. The goal of this work was to identify ORG27569’s binding site at CB1. To this end, we used computational, synthesis, mutation, and functional studies to identify ORG27569’s binding site in the CB1 TMH3-6-7 region. This site is consistent with the results of K3.28192A, F3.36200A, W5.43279A, W6.48356A, and F3.25189A mutation studies, which revealed ORG27569’s binding site overlaps with our previously determined binding site of SR141716A, but extends extracellularly. Additionally, we identified a key electrostatic interaction between ORG27569’s piperidine ring nitrogen and K3.28192 that is important for ORG27569 to act as an inverse agonist. At this allosteric site, ORG27569 promotes an intermediate conformation of the CB1 receptor, explaining ORG27569’s ability to increase CP55,940’s equilibrium binding. This site also explains ORG27569’s ability to antagonize CP55,940’s efficacy in three complimentary ways: (1) ORG27569 sterically blocks movements of the second extracellular loop that have been linked to receptor activation; (2) ORG27569 sterically blocks a key electrostatic interaction between the third extracellular loop residue K373 and D2.63176; and (3) ORG27569 packs against TMH6, sterically hindering movements of this helix that have been shown to be important for receptor activation. My PhD student, Derek Shore, developed the model of the ORG binding site. Compound synthesis was performed by Dr. Herb Seltzman at Research Triangle Institute, mutation studies were performed by Dr. Mary Abood at Temple University and pharmacological studies were performed by Dr. Ruth Ross at University of Toronto.
Identifying Ligands via High-Throughput Screening for the Newly Discovered Cannabinoid Receptor, GPR55.
Work done by Dr. Evangelia Kotsikorou, Post-Doctoral Student and Derek M. Shore, PhD Student
Supported by National Institutes of Health Grants (DA023204 and DA021358)
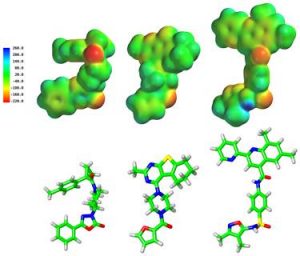 GPR55 is a Class A G protein-coupled receptor (GPCR), highly expressed in human striatum that has been implicated in inflammatory pain, neuropathic pain, metabolic disorder, bone development and cancer. While there is increasing evidence for physiological and pathophysiological roles for GPR55, the paucity of specific antagonists has limited its study. From a β-arrestin, high-throughput, high-content screen of ~300,000 compounds run in collaboration with the Molecular Libraries Probe Production Centers Network initiative, we identified a series of GPR55 antagonists that belong to novel, GPR55 antagonist chemotypes with IC50s in the 0.16 to 2.72 μM range (Heynen-Genel, et al., (2010) “Screening for Selective Ligands for GPR55 – Antagonists” [ML191, ML192, ML193] Bookshelf ID: NBK66153; PMID: 22091481). Importantly, many of the GPR55 antagonists were completely selective, with no observed agonism or antagonism against GPR35, CB1 or CB2 up to 20 μM. By modeling the GPR55 inactive state, we compared the GPR55 binding conformations of a series of antagonists that emerged from this screen. Our modeling indicates that these ligands possess a broad head region that occupies a horizontal binding pocket near the extracellular end of GPR55. This is connected to a central portion that can fit vertically in the receptor binding pocket, terminating in a pendant aromatic or heterocyclic ring that juts out from the central portion of the molecule. It is this pendant ring that renders these ligands, antagonists. Our results will enable second generation GPR55 ligand design and provide a means for distinguishing GPR55 selective ligands from those interacting with cannabinoid receptors. This work was performed in collaboration with Dr. Mary Abood at Temple University, Dr. Marc Caron and Dr. Larry Barak at Duke University, Dr. Thomas Chung at the Sanford-Burnham Institute. Using these initial leads, more potent GPR55 antagonists are being designed at UNCG and synthesized by Dr. Mitch Croatt at UNCG.
GPR55 is a Class A G protein-coupled receptor (GPCR), highly expressed in human striatum that has been implicated in inflammatory pain, neuropathic pain, metabolic disorder, bone development and cancer. While there is increasing evidence for physiological and pathophysiological roles for GPR55, the paucity of specific antagonists has limited its study. From a β-arrestin, high-throughput, high-content screen of ~300,000 compounds run in collaboration with the Molecular Libraries Probe Production Centers Network initiative, we identified a series of GPR55 antagonists that belong to novel, GPR55 antagonist chemotypes with IC50s in the 0.16 to 2.72 μM range (Heynen-Genel, et al., (2010) “Screening for Selective Ligands for GPR55 – Antagonists” [ML191, ML192, ML193] Bookshelf ID: NBK66153; PMID: 22091481). Importantly, many of the GPR55 antagonists were completely selective, with no observed agonism or antagonism against GPR35, CB1 or CB2 up to 20 μM. By modeling the GPR55 inactive state, we compared the GPR55 binding conformations of a series of antagonists that emerged from this screen. Our modeling indicates that these ligands possess a broad head region that occupies a horizontal binding pocket near the extracellular end of GPR55. This is connected to a central portion that can fit vertically in the receptor binding pocket, terminating in a pendant aromatic or heterocyclic ring that juts out from the central portion of the molecule. It is this pendant ring that renders these ligands, antagonists. Our results will enable second generation GPR55 ligand design and provide a means for distinguishing GPR55 selective ligands from those interacting with cannabinoid receptors. This work was performed in collaboration with Dr. Mary Abood at Temple University, Dr. Marc Caron and Dr. Larry Barak at Duke University, Dr. Thomas Chung at the Sanford-Burnham Institute. Using these initial leads, more potent GPR55 antagonists are being designed at UNCG and synthesized by Dr. Mitch Croatt at UNCG.
Recent Publications
J. S. Mnpotra, Z. Qiao, J. Cai, D. L. Lynch, A. Grossfield, N. Leioatts, D. P. Hurst, M.C. Pitman, Z-H. Song, P.H. Reggio. ”Structural Basis of G Protein-Coupled Receptor-Gi Protein Interaction: Formation of the Cannabinoid CB2 Receptor/Gi Complex.” J. Biol. Chem. 289: 20259-20272 (2014). PMID:24855641
D. M. Shore, G. L. Ballie, D. H. Hurst,F. J. Navas, III, H. H. Seltzman, M. E. Abood, R. A. Ross, P. H. Reggio.“Identification of a Putative Allosteric Binding Site at the Cannabinoid 1 (CB1) Receptor.” J. Biol. Chem., 289: 5828-5845 (2014). PMID:24366865
M. Vallée, S. Vitiello, L. Bellocchio, E. H. Chatelain, S. Monlezun, E. Martin-Garcia, G. L. Baillie, F. Panin, A. Cathala, V. Roullot-Lacarrière, S. Fabre, D. P. Hurst , D. L. Lynch, D. M. Shore, V. Deroche-Gamonet, U. Spampinato, J.-M. Revest, R. Maldonado, P. H. Reggio, R. A. Ross, G. Marsicano and P. V. Piazza. “Pregnenolone can protect the brain from cannabis intoxication.” Science, 343(6166) : 94 (2014). PMID:24385629
T. Kimura, K. Vukoti, D.L. Lynch, A. Grossfield, M/C. Pitman, P.H. Reggio, A.A. Yeliseev, K. Gawrisch. “Global Fold of the Human Cannabinoid Type 2 Receptor Probed by Solid-State 13 C-, 15 N-MAS NMR and Molecular Dynamics Simulations.” Proteins, 82(3): 452-465 (2014).PMID:23999926
P. Zhao, T. R. Lane , H. G.L. Gao , D. P. Hurst, E. Kotsikorou, L. Le , P. H. Reggio and M. E. Abood. “Crucial Positively Charged Residues for Ligand Activation of the GPR35 Receptor.” J. Biol. Chem. 289(6): 3625-3638(2014). PMID:24347166
E. Kotsikorou, F. Navas,III, M.J. Roche, A.F. Gilliam, B.F. Thomas, H.H. Seltzman, P. Kumar, Z.H. Song, D.P. Hurst, D.L. Lynch,P.H. Reggio. “The Importance of Hydrogen Bonding and Aromatic Stacking to the Affinity and Efficacy of Cannabinoid Receptor CB2 Antagonist, 5-(4-chloro-3-methylphenyl)-1-[(4-methylphenyl)methyl]-N-[(1S,2S,4R)-1,3,3-trimethylbicyclo[2.2.1]hept-2-yl]-1H-pyrazole-3-carboxamide (SR144528).” J. Med. Chem., 56: 6593-6612 (2013). PMID:23855811
J. Marcu, D. M. Shore, A. Kapur, M. Trznadel, A. Makriyannis, P. H. Reggio and M. E. Abood. “Novel Insights Into CB1 Cannabinoid Receptor Signaling: A Key Interaction Identified Between EC3-Loop and TMH2.” J. Pharmacol. Exp. Ther. 345(2): 189-97 (2013). PMID:23426954
G. Baillie, J. Horswill, S. Anavi-Goffer, P.H. Reggio, M.E. Abood, D. Bolognini, S. McAllister, P.G. Strange, G.J. Stephens, R.G. Pertwee and R.A. Ross. “CB1 Receptor Allosteric Modulaltors Display Both Agonist and Signal Pathway Specificity.” Mol Pharmacol. 83(2): 322-38 (2013). PMID: 23160940
E. Kotsikorou, H. Sharir, D. M. Shore, D. P. Hurst, D. L. Lynch, K. E. Madrigal, S. Heynen-Gene43, L. B. Milan, T. D.Y. Chung, H. H. Seltzman, Y. Bai, M. G. Caron, L. S. Barak, M. P. Croatt, M. E. Abood and P. H. Reggio. “Identification of the GPR55 Antagonist Binding Site Using a Novel Set of High Potency GPR55 Selective Ligands”, Biochemistry 52(52):9456-9469 (2013). PMID: 24274581
J. Cumella, L. Hernández-Folgado, R. Girón, E. Sanchez, P. Morales, D. P. Hurst, M. Gómez-Cañas, M. Gómez, D. C. G. A. Pinto, P. Goya, P. H. Reggio, M. I. Martin, J. Fernández-Ruiz, A.M. S. Silva, and Nadine Jagerovic. “Chromenopyrazoles: Non-psychoactive and Selective CB(1) Cannabinoid Agonists with Peripheral Antinociceptive Properties.” ChemMedChem, 7: 452-463 (2012). PMID:22302767
H. Zheng, E.A. Pearsall, D.P. Hurst, Y. Zhang, J. Chu, Y. Zhou, P.H. Reggio, H.H. Loh, P-Y, Law.“Palmitoylation and Membrane Cholesterol Stabilize µ-Opioid Receptor Homodimerization and G Protein Coupling.” BMC Cell Biol. 13, 6 (March 19), 2012. PMID:22429589





